Abstract
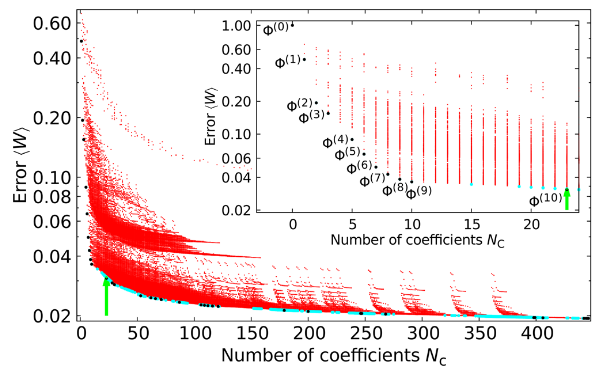 |
Large-scale simulations using interatomic potentials provide deep insight into the processes occurring in solids subject to external perturbations. The atomistic description of laser-induced ultrafast nonthermal phenomena, however, constitutes a particularly difficult case and has so far not been possible on experimentally accessible length scales and timescales because of two main reasons: (i) ab initio simulations are restricted to a very small number of atoms and ultrashort times and (ii) simulations relying on electronic temperature- (Te) dependent interatomic potentials do not reach the necessary ab initio accuracy. Here we develop a self-learning method for constructing Te-dependent interatomic potentials which permit ultralarge-scale atomistic simulations of systems suddenly brought to extreme nonthermal states with density-functional theory (DFT) accuracy. The method always finds the global minimum in the parameter space. We derive a highly accurate analytical Te-dependent interatomic potential Φ(Te) for silicon that yields a remarkably good description of laser-excited and -unexcited Si bulk and Si films. Using Φ(Te) we simulate the laser excitation of Si nanoparticles and find strong damping of their breathing modes due to nonthermal melting. |
Bernd Bauerhenne et al., Self-Learning Method for Construction of Analytical Interatomic Potentials to Describe Laser-Excited Materials, Phys. Rev. Lett.